
20 år med det humane genom – gennem op- og nedture
Det er lidt mere end 20 år siden, at det humane genom for første gang blev kortlagt på DNA-sekvensniveau. Kortlægningen gjorde forskere meget klogere på det at være menneske, men viste også, at vores arvemasse og for eksempel den genetiske risiko for udvikling af sygdom er meget mere kompleks, end man havde troet.
Af Kristian Sjøgren, videnskabsjournalist. ksjoegren@gmail.com
For lidt over 20 år siden færdiggjorde forskere et stykke forskningsarbejde, der af mange blev sammenlignet med det at sende mennesker til Månen.
Overalt i verden havde forskere brugt mere end et årti på at kortlægge hele det humane genom, altså hver en byggesten i det arvemateriale, som gør mennesker til mennesker. Fra projektet startede i 1990, til det blev afsluttet i 2003, arbejdede tusindvis af forskere på at stykke vores arvemateriale sammen bid for bid. Den samlede omkostning løb op i svimlende 2,7 milliarder dollars. I 2001 fremlagde forskerne bag Human Genome Project (HGP) så det foreløbige humane genom. I 2003 blev et mere færdigt genom endelig vist frem for verden. Det er siden blevet forbedret endnu mere.
Drømmen om at sekventere det humane genom var allerede opstået tilbage i 1950’erne, da den amerikanske biolog James Watson og den engelske fysiker Francis Crick opdagede, at DNAet i vores arvemateriale har en dobbelt helix-struktur. To andre knapt så kendte forskere havde også del i opdagelsen – fysikeren Maurice Wilkins samt kemikeren Rosalind Franklin.
Med det samme opstod der er brændende ønske om at kunne zoome ind på kromosomernes DNA og kortlægge hver en bid af arvematerialet. Tanken var, at vi med bedre forståelse af, hvordan vi mennesker genetisk er skruet sammen, også bedre kunne forstå alt fra vores forhistorie til baggrunden for udvikling af arvelige sygdomme.
Én af de forskere, som har arbejdet med det humane genom gennem de mange årtier, er den danske professor og pionér indenfor bioinformatik Søren Brunak fra Novo Nordisk Foundation Center for Protein Research ved Københavns Universitet. Han fortæller, at kortlægningen af det humane genom ikke bare har givet unik indsigt i, hvad det vil sige at være menneske, men også hvor komplekse vi er som organismer.
»Da man satte projektet i gang i 1990, spekulerede mange af verdens førende forskere blandt andet i, hvor mange proteinkodende gener der ville være i det humane genom. Den almindelige antagelse var, at der måtte være omkring 150.000, fordi det var nogenlunde det, der var plads til, med det man vidste om længden på generne. Det viste sig, at vores arvemateriale faktisk kun indeholder 21.000 proteinkodende gener. Det var en kæmpe overraskelse, som gjorde det nødvendigt at gentænke alt, hvad vi troede om generne, og hvordan de fungerer,« fortæller Søren Brunak.


Blandede DNA fra flere personer
Går vi tilbage til 1990 og drømmen om at kortlægge det humane genom, var alle klar over, at det ville blive en opgave, der ville komme til at tage en menneskealder. Simpelthen fordi man på daværende tidspunkt ikke havde metoder til at kortlægge arvematerialet hurtigere. Man håbede dog på, at der med tiden ville blive udviklet metoder, som kunne sætte fut i forskningen. Ydermere var projektet kendetegnet ved at være eksplorativt i modsætning til hypotesedrevet. Det vil sige, at forskerne “bare” ville udforske det humane genom uden at vide, hvad de ville finde. Desuden havde projektet sit udgangspunkt i akademia. Det var med andre ord ikke meningen, at det skulle finansieres af industrien – i hvert fald ikke til at starte med.
»Der var lidt Columbus-håb over det. Normalt er det god latin at have en hypotese, inden man kaster sig ud i et stort medicinsk forskningsprojekt, men her gjorde man det omvendt og indsamlede enorme mængder data, før man begyndte at formulere hypoteserne. Det store mål var dog at etablere et humant referencegenom, som forskningen indenfor feltet kunne sammenligne sig op imod, når man stod med DNA fra et enkelt menneske,« forklarer Søren Brunak.
Han uddyber, at det desuden blev besluttet, at man i stedet for at kortlægge én persons arvemateriale faktisk blandede DNA fra flere personer sammen. Opgaven blev så sendt rundt i verden, hvor en forskergruppe kunne få til opgave at kortlægge et af menneskets 23 kromosomer, mens andre måske blot skulle kortlægge en lille del af et andet. I tildelingen af kromosomer blev der af konsortiet taget hensyn til, at nogle forskere havde særlig interesse i for eksempel sygdomme associeret til det ene eller andet kromosom.
»Teknologisk er vi et helt andet sted i dag, hvor vi indenfor én dag kan kortlægge et individs genom. Vi er også meget mere interesserede i at finde ud af, hvad der i dit DNA gør dig til dig, og hvad der i mit DNA gør mig til mig. Det har meget større relevans for blandt andet sygdomsforståelse og individualiseret behandling. Dengang ønskede man dog i første omgang blot det store “søkort”, og så måtte man zoome ind på individet senere,« siger Søren Brunak.
Fra møjsommelig teknologi til hurtig shotgun-sekventering
Fra møjsommelig teknologi til hurtig shotgun-sekventering
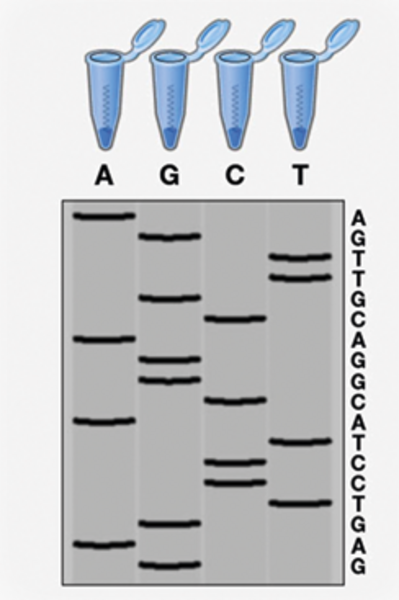
Den oprindelige metode til at kortlægge DNA blev opfundet af den britiske biokemiker Frederick Sanger i 1977 og blev også opkaldt efter opfinderen – Sanger-sekventering. Sanger-sekventering var i årtier den foretrukne metode til at afkode sekvensen i en DNA-streng, hvilket metoden også kunne gøre med næsten 100 procents præcision.
Kort fortalt fungerer Sanger-sekventering på den måde, at forskere kopierer det DNA, som de ønsker at kende sekvensen på. Når en DNA-sekvens skal kopieres, skal der på kopien påsættes DNA-byggesten i form af nukleotider én efter én. Der findes fire forskellige nukleotider, og efterhånden som de bliver sat på, bliver DNA-strengen længere og længere. Forskere kan sortere de forskellige længder i en gel og kan med en laser måle, hvilken byggesten der sættes på som næste trin i kopieringen. Det kan de gøre ved brug af blandt andet nukleotider, der er markeret med et fluorescerende protein.
Til sidst kan forskerne stykke den fulde DNA-streng sammen og præsentere den samlede genetiske kode. (Se figuren til højre). Denne metode er særligt velegnet til DNA-strenge, som er omkring 500-1000 nukleotider lange. I det humane genom findes der tre milliarder byggesten.
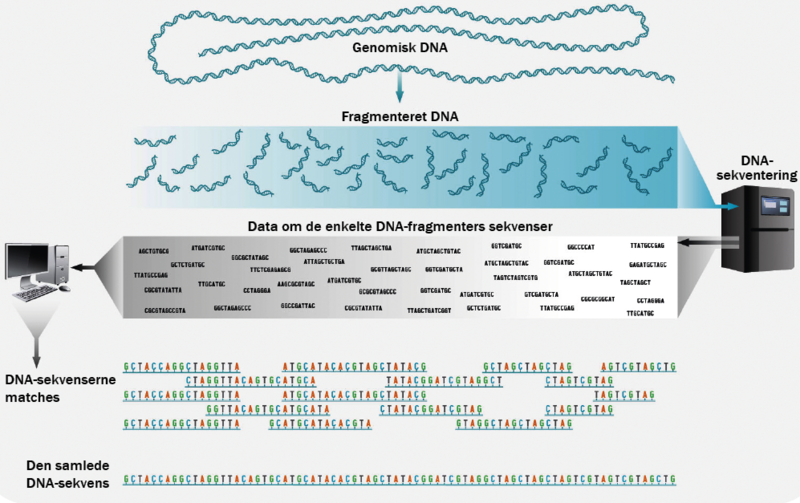
I moderne DNA-sekventering har man paralleliseret proceduren. I stedet for at kortlægge ét stykke DNA fra den ene ende til den anden, skærer man ved helgenom shotgun-sekventering alt DNA’et i millioner af små stumper på mellem 100 og 1.000 nukleotider.
Hvert tilfældigt udvalgte område i DNA’et bliver derefter sekventeret mange gange samtidigt. Ved hjælp af computeralgoritmer kan man efterfølgende sammensætte DNA-stumperne til hele DNA-strenge igen, som det fremgår af figuren nedenfor:
Entreprenør kommercialiserede det humane genom
Selvom det humane genomprojekt indledningsvist var tænkt som et ikke-kommercielt anliggende, gik der ikke mange år, før industrien også begyndte at fatte interesse i projektet. Én person, som kom til at have stor betydning for, at det humane genom blev kortlagt langt hurtigere, end man troede, var den amerikanske bioteknolog og forretningsmand Craig Venter.
Craig Venter så en forretningsidé i at sætte fut i projektet og være med til at opdage gener, der koder for proteiner. Han tænkte, at de nyopdagede, proteinkodende gener kunne patenteres, og at der derfor kunne tjenes penge på dem.
For at realisere denne drøm var der dog behov for, at tingene fik mere fart på, så Craig Venter udviklede en metode til meget hurtigere at sekventere hele arvematerialet. I stedet for at sekventere DNA’et bid for bid klippede han hele det humane DNA i småstykker i en molekylær blender og sekventerede det hele på én gang. Derefter fik han bioinformatikere til at udvikle sofistikerede analytiske værktøjer til at sætte DNA’et sammen igen.
»Mange mente dengang, at det var totalt urealistisk, fordi de mange stumper passede sammen på for mange forskellige måder. Det svarede til at skulle samle millioner af blå puslespilsbrikker til ét puslespil af en himmel. Men det lykkedes bioinformatikerne at skabe dette værktøj. For Craig Venter betød det, at han kunne komme tættere på målet om at kortlægge det humane genom og gøre data tilgængelige for virksomheder mod betaling,« siger Søren Brunak.
Trods Craig Venters fremsynethed, når det kommer til metoder til at kortlægge det humane genom, havde han ikke det samme fremsyn i sin forretning, der havde mere end almindeligt svært ved at tjene nok penge til at dække de enorme omkostninger ved at kortlægge det humane genom.
»En sjov sidehistorie er, at mens hele verden forsøgte at kortlægge et referencegenom, som var en blanding af flere forskellige personers DNA, benyttede Craig Venter sit eget genom i hans projekt. Han var derfor også den første person til at få kortlagt sit eget genom og hævdede at kunne ændre livsstil på baggrund af fundene i genomet,« fortæller Søren Brunak.
Sygdom skyldes sjældent enkelte gener
Spoler vi tiden frem til 2003, kom den daværende amerikanske præsident Bill Clinton på forsiden af det videnskabelige magasin Science i forbindelse med offentliggørelsen af det humane referencegenom. Ved hans ene side stod Francis Collins, der havde været leder af HGP, der repræsenterede det offentlige projekt, og på hans anden side stod Craig Venter.
Med den endelige kortlægning af det humane genom var startskuddet gået til nu to årtiers forskning i, hvad den viden, som ligger i det humane genom, kan bruges til. Nærliggende var det selvfølgelig at tro, at man nu hurtigt kunne finde generne for tusindvis af sygdomme fra diabetes og demens til hjertekarsygdomme og sklerose. Det viste sig dog, at den gamle idé og håb om “ét gen til én sygdom” havde være overoptimistisk og en forsimpling. Havde der været de forventede 150.000 gener, kunne man måske have forestillet sig, at specifikke gener var relateret til specifikke sygdomme, men med kun 21.000 humane gener blev billedet meget mere komplekst.
»Fordi der er så få proteinkodende gener, ser det ud til, at effekten på det, som man kalder komplekse sygdomme, ofte er spredt ud over mange hundrede gener. For eksempel kan en højere risiko for udvikling af diabetes være resultatet af små variationer i måske 400 eller 500 gener. Det er meget indviklet at finde ud af, hvordan varianter i så mange gener spiller sammen om at komme til udtryk som sygdomsrisiko. Det havde da været nemmere, hvis vi bare kunne pege på ét gen som årsagen til en sygdom og sætte ind med behandlinger mod dette ene gen, men sådan er det ikke. Generne påvirker hinanden og arbejder sammen. Derfor blev mange også skuffede over, at kortlægningen af det humane genom ikke med det samme ledte til udvikling af en masse lægemidler mod en lang række store sygdomme,« forklarer Søren Brunak.
Erkendelsen af kompleksiteten ledte dog til udvikling et helt nyt forskningsfelt, systembiologien, som netop er Søren Brunaks speciale. Systembiologi går blandt andet ud på at finde ud af, hvordan generne samarbejder, og udnytte regnekraften fra meget store computere til at hitte rede i store datasæt. Blandt andet prøver man finde ud af, hvad varianter i gener samlet set betyder for risiko for udvikling af sygdom, og om vi kan udregne en persons sygdomsrisiko bare ved at kigge på varianter i hundredvis af gener.
Satte gang i guldfeber
Med kortlægningen af det humane genom kunne forskere også for første gang se, at vores arvemasse består af meget, meget mere end bare proteinkodende sekvenser. Blandt andet består vores DNA af tusindvis af gener, der aldrig bliver oversat til proteiner, men i stedet bliver oversat til små stumper af RNA, der kan regulere mange andre gener ved at binde til dem.
»Det blev hurtigt en af de store guldgraverhistorier, fordi det åbnede op for muligheden for at identificere RNA-gener med betydning for risikoen for udvikling af for eksempel kræft. RNA-generne spiller en stor rolle og har indflydelse på rigtig mange sygdomme og er også interessante mål ved behandling af sygdomme. Disse gener blev også startskuddet på et “goldrush”, og det er det stadig i dag,« siger Søren Brunak.
Han fortæller, at der i dag også er meget fokus på områder af genomet, der koder for små peptider, som man kan opfatte som mikroproteiner. Der findes mange tusinder mikroproteiner, som bliver udtrykt af vores arvemasse, og som vi for en stor dels vedkommende endnu ikke kender funktionen af.
»Et gen kan være rigtig mange ting, og der kan komme mange ting ud af et gen. Et gen kan kode for et protein, en RNA-stump eller et peptid, men de små proteiner kan også være et nedbrydningsprodukt, når større proteiner med en given funktion bliver nedbrudt til et nyt molekyle med en anden funktion. Vi er slet ikke færdige med at opdage nye ting, og det humane referencegenom viste os, hvor komplekst det hele er,« siger Søren Brunak.
»Det er ikke altid nødvendigt at have en hypotese på forhånd og slet ikke, hvis man skal finde det helt uventede«, siger han.
Junk-DNA er ikke skrald
Gennem de seneste 20 år er forskere også blevet klogere på noget i vores arvemateriale, der optog rigtig mange mennesker før i tiden. Her taler vi om såkaldt “junk-DNA”. Allerede tilbage omkring 1990 var mange forskere skeptiske over for ideen om at kortlægge hele det humane genom, idet det allerede dengang var velkendt, at ikke hele genomet koder for proteiner. Når en hel del arvematerialet således blev anset for ligegyldigt skrald, gav det ikke mening at bruge tid på at kortlægge dette.
Junk-DNA er i store træk betegnelsen for genetisk information, der ikke bliver benyttet mere. Forestil dig, at en forfader millioner af år tilbage i tiden har produceret et protein, som for eksempel var nødvendigt for at lave en tyk og varm pels. Ja, det gen har vi jo ikke rigtig brug for mere, så det er der blevet lukket ned for i vores arvemateriale. Over tid er genet blevet muteret igen og igen, indtil det i dag blot er en samling DNA-stumper uden funktion. Det er junk-DNA.
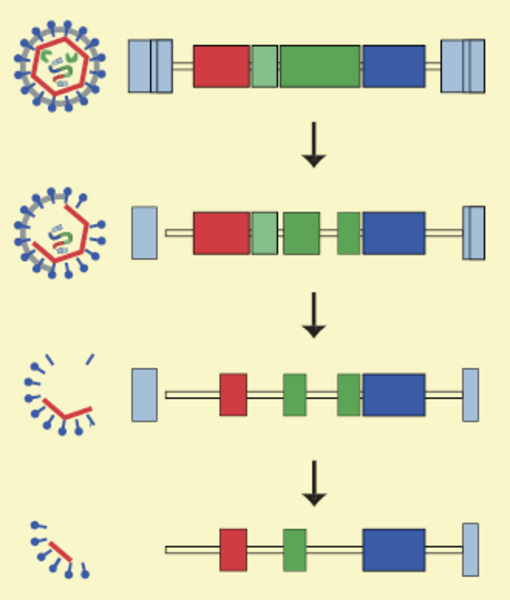
En del af det DNA i vores arvemasse, vi i dag kalder junk-DNA, stammer faktisk oprindelig fra virus, der har kopieret deres arvemasse ind i vores forfædres DNA.
Figuren viser, hvordan sådant virus-DNA med tiden nedbrydes, så der ikke længere kan produceres hele viruspartikler. Men enkelte gener kan godt være intakte, så der stadig produceres virusproteiner.
Illustration: Palle Villesen.
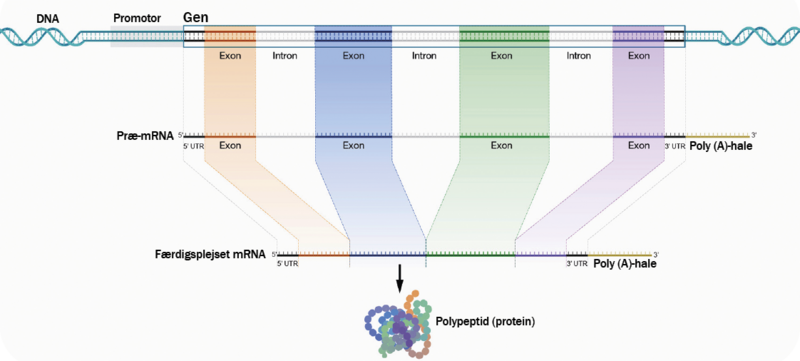
Tanken om, at kun nogle få procent af vores arvemateriale er nyttigt DNA, er dog helt forkert. Selvom en stor del af selve generne i sig selv indeholder dette ikke-proteinkodende DNA, er det ofte ingenlunde uden funktion. Et gen består af både kodende dele (kaldet exons) og ikke-kodende dele (kaldet introns), og begge dele af genet spiller en vigtig rolle for, hvordan et gen bliver udtrykt, reguleret, oversat til protein og finder sin funktion.
»Så selv den ikke-kodende del af et gen er vigtig og vigtig at forstå og kortlægge. Hvis man i en organisme fjernede alle de ikke-proteinkodende dele af et gen, ville der ikke komme noget godt ud af det. For eksempel sidder mange af de RNA-kodende gener i introns,« siger Søren Brunak.
Han uddyber, at evolutionen ikke fungerer på den måde, at arvematerialet bare nulstilles, når der ikke længere er brug for et gen. Alt det, som nogle kalder for junk-DNA, kan man i stedet opfatte som gamle “genetiske projekter”, der måske ikke bliver benyttet til den funktion, de oprindeligt havde, men som i kraft af deres tilstedeværelse stadig kan spille en rolle i hele det genetiske udtryk.
»Tilbage i 1980'erne omskrev den japanske forsker Ohno byggestenene i DNA’et til noder og viste, at man faktisk kan lytte sig frem til disse områder med junk-DNA. I selve de kodende dele af et gen foregår det hele rytmisk, fordi genet bliver afkodet tre byggesten ad gangen. I junk-delene er det hele mere rodet, og det kan man direkte høre,« fortæller Søren Brunak.
Behov for genomprofil på millioner af mennesker
Spoler vi tiden frem til 2024, er forskere for længst kommet sig over, at de ikke kan relatere ét gen til én sygdom, i hvert fald for mange af de store folkesygdomme. Nogle sygdomme er resultatet af ganske få genetiske varianter, men flere af de store sygdomme, blandt andet type 2-diabetes, hjertekarsygdomme og svær overvægt, opstår oftest i et meget komplekst samspil mellem generne. Det betyder også, at for at kunne kortlægge dette komplicerede samspil har forskere brug for millioner af patientjournaler på millioner af mennesker, som alle sammen har fået kortlagt deres DNA.
Søren Brunak arbejder selv med netop disse ekstremt store datasæt, hvor han med sine kollegaer prøver at forstå genetikken bag blandt andet udvikling af type 2-diabetes. Det er dog slet ikke muligt for et menneske at hitte hoved og hale i så gigantiske datasæt, så det må forskerne have supercomputere og kunstig intelligens til at gøre for sig. Til gengæld kan de så også få et vindue ind til genetikken bag sygdom og få en bedre forståelse af sygdomsmekanismer.
»På den måde kan vi se, hvad der genetisk leder til udvikling af type 2-diabetes, og også hvordan de biologiske mekanismer, som leder til sygdommen, kan være forskellige fra menneske til menneske. Det samme gælder også en sygdom som demens,« siger Søren Brunak.
Han uddyber, at denne indsigt ikke havde været mulig, hvis forskere ikke havde taget skridtet videre fra det humane referencegenom til at kortlægge millioner af menneskers individuelle genomer.
»Og så manglede man pludseligt sygdomsdata for enkelte patienter, for det havde man ikke sat gang i i 1990. Referencegenomet var et vigtigt udgangspunkt for at forstå human genetik, men i dag sammenholder vi i stedet den enkeltes genetik med for eksempel sygehistorik, men også andre træk, for eksempel levevis, tillidsfuldhed eller andre bløde karakteristika,« siger Søren Brunak.
Det humane genom har også gjort os klogere på menneskets fortid
Det humane genom har også gjort os klogere på menneskets fortid
Foruden af gøre os klogere på sundhed og sygdomme har kortlægningen af det humane genom også gjort os klogere på menneskets fortid. Med kortlægningen af det humane genom blev det nemlig muligt at se, hvordan vi har udviklet os over forhistorisk tid, og blandt andet opdage, hvor forskellige vi er fra vores nærmeste slægtninge, neandertalerne, men også at vi mennesker faktisk har blandet gener med neandertalerne på et tidspunkt i forhistorien. Derfor er op imod to procent af europæernes DNA neandertaler-DNA.
Kortlægningen af det humane genom har også gjort det muligt at kigge på, hvordan befolkninger i Europa har ændret sig over tid. De seneste undersøgelser fra professor Eske Willerslev og hans kolleger på Københavns Universitet har vist, at de oprindelige europæiske jægersamlere, der levede i Europa for 10.000 år siden, ikke udviklede sig til landbrugsfolk, men faktisk blev erstattet af et folk, som kom med landbruget fra Mellemøsten. Der var dog en vis grad af assimilation, hvilket betyder, at europæere i dag har varierende grad af jæger-samler-DNA i deres arvemasse. Folk fra Sydeuropa har mere jæger-samler-DNA i deres arvemateriale end folk fra Nordeuropa. Landbrugsfolket blev dog også selv erstattet for mellem 5.000 og 6.000 år siden af et hyrdefolk fra de store sletter i Ukraine og Rusland. Dette hyrdefolk, som hed yamnayaerne, kom til Europa med store flokke af dyr og erstattede på meget kort tid det folk som levede her.
I dag har europæerne også varierende grad af Yamnaya-DNA i deres arvemateriale. I Nordeuropa er andelen af yamnaya-DNA størst, og det betyder faktisk, at vi også har den største risiko for udvikling af blandt andet multipel sklerose. Generne for multipel sklerose stammer nemlig fra yamnayaerne. I Sydeuropa, hvor folk har mere jæger-samler-DNA i deres arvemateriale og mindre yamnaya-DNA, er forekomsten af multipel sklerose mindre.
Gener og teknologi bliver en del af lægens værktøjer
Kigger vi ind i fremtiden for at se, hvad det humane genomprojekt kan få af betydning over 30 år, 40 år eller på endnu længere sigt, ser Søren Brunak stadig et stort behov for at forstå, hvordan genetisk disposition leder til udvikling af sygdomme under paraplybetegnelsens “store folkesygdomme”. Det drejer sig om diabetes, KOL, forhøjet kolesteroltal, forhøjet blodtryk osv. og herunder, hvordan det enkelte menneske udsættes for miljøpåvirkninger.
Derudover ser professoren også store perspektiver i meget hurtigere at kunne kortlægge en persons genetik og bruge viden fra genomet til at lave hurtige interventioner. Han forestiller sig som eksempel, at det, der i 1990’erne og op gennem 00’erne tog 13 år at kortlægge, bliver kogt ned til få timer, så et nyfødt barn kan få lavet en genprofilering ud fra den dråbe blod , som bliver taget i den såkaldte hælprøve. Her vil man lynhurtigt kunne se, om barnet har en genetisk disposition for alvorlig sygdom, som man kan gøre noget ved. Det kan som eksempel være, at barnet ikke kan producere et givent protein, som er nødvendigt for at overleve eller udvikle sig korrekt. Med denne viden allerede på fødselstidspunktet kan læger intervenere og behandle med det relevante protein, så barnet aldrig når at blive alvorligt syg.
»Mange nyfødte børn ender med at cykle rundt i systemet i mange år, men det kan undgås, hvis man fra fødslen får kortlagt, om de har en given sygdom på grund af for eksempel en gendefekt. I Danmark har man med Nationalt Genom Center besluttet at gøre denne information let tilgængelig for lægerne. Læger er magikere, men de kan ikke holde styr på genetikken bag tusindvis af sygdomme. Det skal gøres elektronisk, så lægerne får et støtteværktøj, der kan hjælpe med at komme med mere præcise diagnoser på baggrund af genetiske data. Indenfor den kommende tid vil vi se, at læger i stigende grad bliver støttet af værktøjer, der kan fortælle en hel masse om en persons sygdom eller sygdomsrisiko, og ikke blot finde årsagen til sygdom, men også forebygge den,« siger Søren Brunak. ♦

Søren Brunak er professor ved Københavns Universitet, Novo Nordisk Foundation, Center for Protein Research.