
Kunstig intelligens baner vejen for fremtidens katalytiske materialer
Udviklingen indenfor kunstig intelligens har åbnet døren for, at materialeforskere kan forudsige nye materialer med nyttige egenskaber. Neurale netværk hjælper os således i dag med at finde nye katalytiske materialer, som er vigtige for en bæredygtig fremtid.
Af Mie Andersen & Nikolaj Rønne
Katalytiske materialer spiller en afgørende rolle i industrien ved at fremskynde kemiske reaktioner. De bruges til alt fra fremstilling af medicin til produktion af ammoniak til landbruget.
Men vi står nu over for en ny udfordring: At finde katalytiske materialer, der kan hjælpe med at med at opnå en mere bæredygtig produktion og reducere vores klimaaftryk. Nye og mere effektive katalytiske materialer er afgørende, fordi de både kan sænke energiomkostningerne for de industrielle processer og reducere CO2-udledningen markant.
Moderne kunstig intelligens (AI), især neurale netværk, har revolutioneret måden, vi kan forudsige og udvikle nye materialer på. Traditionelt kræver det enorme mængder af computerkraft at forudsige stabiliteten og egenskaberne af et nyt katalytisk materiale. Neurale netværk kan erstatte disse komplekse beregninger og give os svarene langt hurtigere.
I stedet for at gennemføre computertunge, kvantemekaniske beregninger for at vurdere, om et materiale er stabilt, eller om det er en god katalysator, kan AI-modeller nu forudsige disse egenskaber ud fra tidligere data. Det betyder, at vi kan undersøge mange flere materialer på kortere tid.
En anden mulighed er at bruge AI-modeller til at foreslå specifikke materialer med ønskede egenskaber – for eksempel en katalysator, der virker effektivt ved lavere temperaturer. Dette kaldes generativ AI, og det har vi for nyligt opnået lovende resultater med. Denne nye mulighed for at designe materialer “fra bunden” giver os adgang til at udforske tusindvis af potentielle nye katalysatorer. Vi forventer derfor, at vi de kommende år vil se en rivende udvikling af nye materialer baseret på brug af generativ AI.
Sammenlagt betyder disse fremskridt, at vi nu kan undersøge og udvikle langt flere materialer end tidligere. Det giver håb om at finde nye og mere effektive katalysatorer, der kan hjælpe industrien med at mindske sit klimaaftryk.
Nobelprisen i fysik 2024
Nobelprisen i fysik 2024


Nobelprisen i fysik 2024 er givet til John J. Hopfield og Geoffrey E. Hinton for deres banebrydende opdagelser indenfor neurale netværk – teknologier, der har lagt fundamentet for kunstig intelligens som ChatGPT.
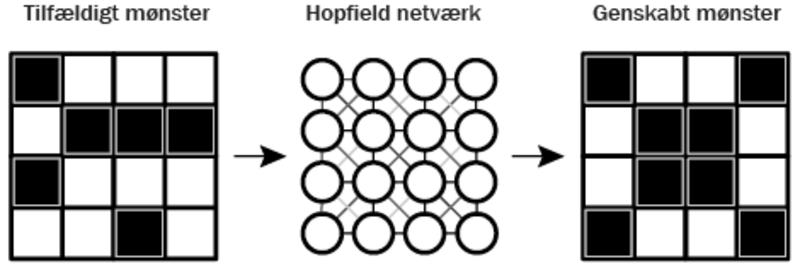
John J. Hopfield udviklede Hopfield-netværket – en metode til at skabe computerhukommelse inspireret af, hvordan menneskets hjerne fungerer. Netværket bruger såkaldte neuroner, små enheder, der alle er forbundet med hinanden for at lære og gemme information. Hopfield-netværket er inspireret af fysikkens måde at beskrive magnetiske materialer på. Ligesom små magneter i et materiale kan finde et mønster, hvor de for eksempel alle peger i samme retning, kan neuronerne i et Hopfield-netværk finde og huske mønstre i data. Netværket “lærer” derfor at gemme information på samme måde, som magneter kan danne stabile mønstre i et magnetisk materiale.
Geoffrey E. Hinton, ofte kaldet “den kunstige intelligens’ fader”, populariserede den metode, vi bruger til at træne neurale netværk i dag – kaldet “backpropagation”. Han udviklede også Boltzmann-maskinen, et neuralt netværk, der kan bruges til at genkende mønstre og klassificere data. Boltzmann-maskinen er inspireret af ideer fra statistisk fysik og fungerer ved at finde mønstre i data på samme måde, som partikler i et materiale søger mod de mest stabile tilstande. Ligesom partikler tiltrækkes af lavere energi, justerer maskinen sine forbindelser for at finde de mest sandsynlige mønstre – en proces, der hjælper med at genkende komplekse mønstre i data.
Forudsigelse af katalytiske egenskaber
Hvordan kan man (op)finde et nyt og bedre materiale? Traditionelt har eneste redskab til rådighed været omstændelige laboratorieforsøg, hvor man ved “trial and error” fremstiller og tester forskellige mulige materialer.
Et godt eksempel er Thomas Edison, som siges at have testet omkring 1600 forskellige materialer under udviklingen af glødepæren i 1870’erne. Samspillet mellem teori, kvantemekaniske beregninger og AI-modeller kan fremskynde denne proces ved at foreslå nye materialer med specifikke egenskaber, så krævende laboratorieforsøg kan reserveres til kun de allermest lovende materialer.
Når vi skal forstå og forudsige katalytiske egenskaber af et materiale, er det vigtigt at forstå, hvor godt molekyler binder sig til materialets overflade. Energien, der kræves for at adskille molekylet fra overfladen – bindingsenergien – er en afgørende parameter for, hvor god katalysatoren er. Bindingsenergien kan beregnes ved hjælp af kvantemekanikkens komplekse ligninger.
Men der er et problem: Disse beregninger er utroligt tidskrævende på trods af, at vi i dag har adgang til supercomputere. Det skyldes, at beregningstiden vokser voldsomt med antallet af elektroner i materialet. Hvis det for eksempel tager en dag at regne på et materiale med 1000 elektroner, ja så vil beregningen på et materiale med det dobbelte antal elektroner typisk tage otte gange så lang tid – det vil sige otte dage! Ydermere kræver hvert materiale typisk mange forskellige beregninger, hvilket gør det umuligt at undersøge ret mange materialer.
Den moderne vej til materialeinnovation
Det er her AI kommer ind i billedet. En AI-model kan forudsige bindingsenergier på en brøkdel af den tid, vi skal bruge med traditionelle metoder. Men, der er også en hage ved historien. For at virke godt, skal AI-modellen først trænes på data for kendte materialeegenskaber. Og disse data skal stadig beregnes på supercomputere. Derfor er det afgørende, at AI-modellen kan udnytte informationen gemt i data optimalt og lære mest muligt fra så lidt data som muligt.
I vores forskning har vi blandt andet arbejdet på at udvikle nye AI-modeller, der er bedre til at lære, hvordan komplekse molekyler binder til overflader. En vigtig indsigt, vi har fået under dette arbejde, er, at det er afgørende, hvordan de enkelte atomer er placeret geometrisk i molekylet, og hvordan hvert atom er bundet til molekylets andre atomer og til atomerne i katalysatoroverfladen.
Ved at benytte matematisk grafteori kan vi give vores AI-model disse informationer og få den til at bruge dem effektivt. I grafen er hvert atom et “knudepunkt” med specifikke egenskaber, der afhænger af atom-typen, og bindingerne mellem atomer er “forbindelser”.
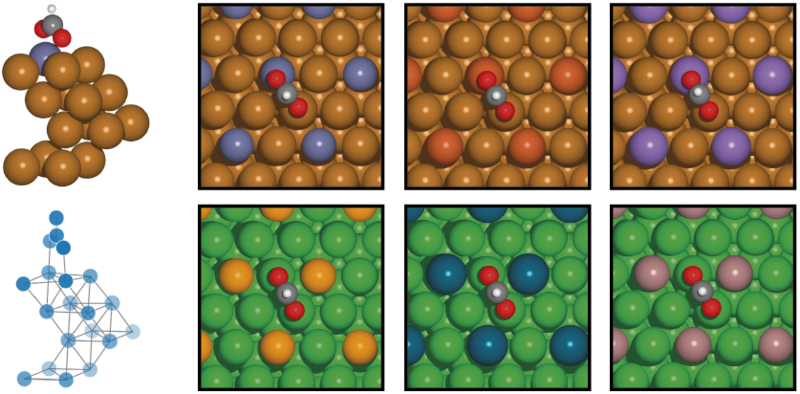
Venstre: Den geometriske struktur (øverst) og strukturen repræsenteret som en matematisk graf (nederst) for molekylet format (HCO2) på en kobber-overflade iblandet zink.
Højre: Format på forskellige materialeoverflader dannet ved at blande elementer fra det periodiske system: Zink, jern og mangan i en kobber-overflade (øverst) samt selen, palladium og gallium i en nikkel-overflade (nederst). Finjustering af materiale-kompositionen gør det muligt at justere vigtige parametre som bindingsenergier af molekyler til overfladen. Illustration: Mie Andersen.
AI-modellen kan forudsige bindingsenergier af nye kombinationer af molekyler og overflader, for eksempel overflader dannet ved at blande forskellige elementer fra det periodiske system. Den kan dermed hjælpe til med hurtigt at afgøre, hvorvidt et nyt materiale er så lovende, at det er værd at investere dyre laboratorieforsøg på at undersøge det nærmere.
AI for materialer
Kan AI-modeller også bruges til andet end at forudsige bindingsenergier?
Ja, faktisk kan vi lære dem at forudsige næsten alle tænkelige materialeegenskaber, inklusiv den totale energi af strukturer, vi kunne finde på at bygge ud fra elementerne i det periodiske system. Det vil være nyttigt i mange andre sammenhænge.
Som et eksempel: Inden vi begynder at regne på, om en given overflade er en god katalysator, ja så skal vi først undersøge, om overfladen overhovedet er stabil nok til, at det vil være muligt at syntetisere den i et laboratorie. I naturen eller i laboratoriet dannes materialer spontant i strukturer med lav energi, så stabile strukturer er her strukturer, der har en lavere energi end andre mulige strukturer. Også i denne sammenhæng har det vist sig nyttigt at beskrive materialer som grafer.
I et såkaldt graf-neuralt netværk lærer AI-modellen, hvordan atomer i grafen vekselvirker med sine naboer. Netværket lærer ved, at hvert atom “snakker” med sine naboatomer og udveksler information om, hvilke typer atomer de er, og hvordan de er forbundet. Forestil dig, at hvert atom sender beskeder om sine egne egenskaber til de atomer, det er bundet til, og får beskeder tilbage om deres. Denne udveksling sker flere gange, og AI-modellen lærer dermed at forstå, hvordan atomerne interagerer i materialet som helhed.
Når AI-modellen først har lært disse mønstre, kan den forudsige energien for nye, ukendte materialer, næsten som om den har sin egen indbyggede fysikforståelse. Idet vi her undgår at tage hensyn til alle de detaljer, der normalt kræves i kvantemekaniske beregninger, kan modellen gøre arbejdet langt hurtigere. Og for at sikre, at AI-modellen rammer plet, tester vi den på nye materialer og sammenligner resultaterne med de traditionelle kvantemekaniske beregninger. Det viser sig ofte, at AI-modellen kan være lige så nøjagtig som de traditionelle metoder.
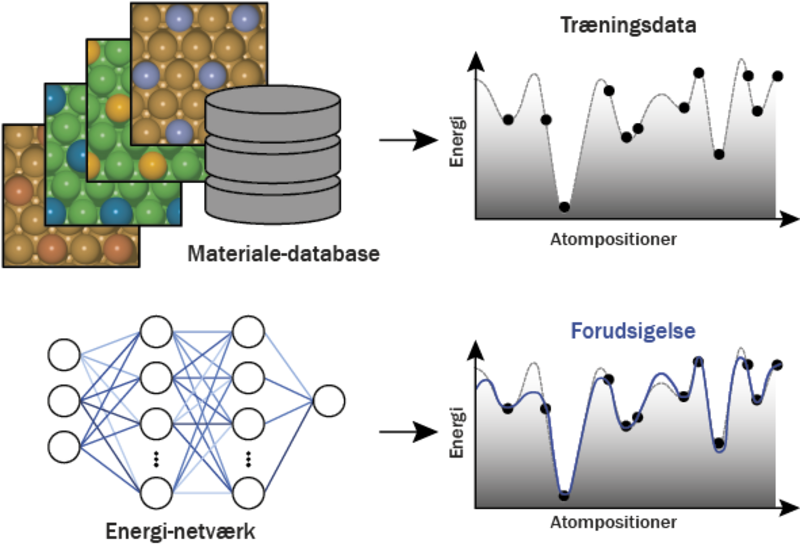
En database af materialer med tilhørende kvantemekaniske energier beskriver delvis energi-funktionen for materialerne. Energi-netværket kan lære at forudsige energien af materialerne på en brøkdel af den tid, som skulle bruges for at beregne den kvantemekaniske energi. Illustration: Nikolaj Rønne
Fra forudsigelse til opdagelse
Men lad os gå et skridt videre. AI kan ikke kun hjælpe os med at forudsige energierne af kendte materialer – den kan også være med til at opfinde nye! Ligesom du kan bede en AI-model om at lave et billede af en by i fremtiden eller et portræt i en bestemt stil, kan vi bruge generativ AI til at skabe materialer med ønskede egenskaber.
I forbindelse med et forskningsophold på University of Toronto, hvor også Nobelprismodtager Geoffrey E. Hinton er professor, har vi udviklet generativ AI til ikke blot at kunne generere billeder, men med samme grundlæggende teknologi, at kunne opfinde nye materialer. Den metode, vi har udviklet, er en såkaldt diffusionsmodel for materialer, som lærer at “rydde op” i materialernes struktur og optimere atomerne, så de placeres på bedre måder. Det kunne være måder, der giver en lavere energi – eller i det mere generelle tilfælde – måder, der forbedrer egenskaberne af materialet.
Forestil dig, at vi starter med et materiale, hvor atomernes placering er lidt tilfældig, næsten som om vi ser på et sløret billede. Diffusionsmodellen lærer at “fjerne støjen” ved gradvist at ændre atomernes placering, så de ender i en bedre struktur. Processen kan minde om at rydde op i et rod – modellen arbejder trin for trin og flytter atomer rundt, indtil de står i den mest fordelagtige konfiguration.
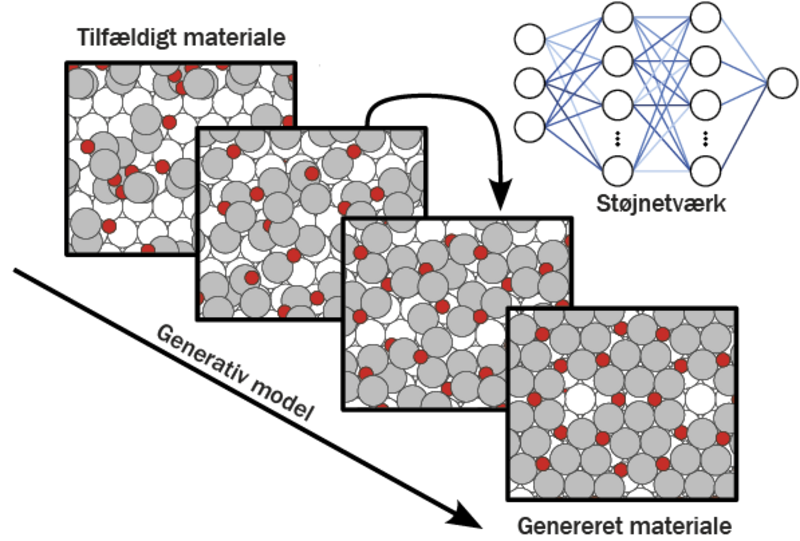
For at træne sådanne diffusionsmodeller bruger vi store mængder data om materialer, der ligner dem, vi ønsker at designe. Ud fra disse eksempler lærer AI-modellen at foreslå kombinationer og strukturer, som mennesker måske aldrig ville have tænkt på. Og det går langt hurtigere, end hvis vi skulle prøve os frem i laboratoriet.
AI-modellen kan nemlig både foreslå nye materialer og teste deres egenskaber ved hjælp af andre AI-modeller. På den måde kan vi afprøve langt flere materialer, end det tidligere var muligt. Og vi kan opdage nye løsninger, som kan ændre måden, vi udvikler katalytiske materialer på.
AI gør det muligt at tage et kæmpe spring fremad i materialeforskningen. Hvad vi tidligere brugte måneder – måske endda år – på at beregne og afprøve, kan nu gøres på få øjeblikke. Det åbner døren for en helt ny æra indenfor udviklingen af materialer med præcis de egenskaber, vi drømmer om. ♦
Neurale netværk
Neurale netværk
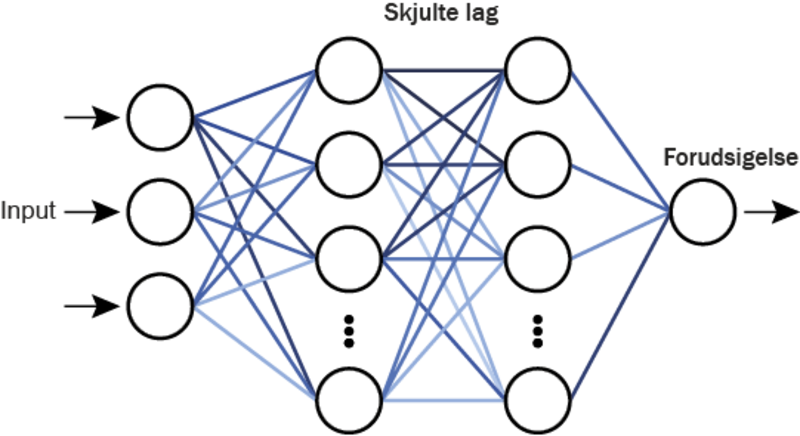
Et neuralt netværk er hjertet i kunstig intelligens. Det er denne teknologi, der gør det muligt for AI at lære, genkende og huske mønstre. I sin enkleste form består et neuralt netværk af mange små enheder, kaldet neuroner, som er forbundet med hinanden. Du kan tænke på en neuron som en lille kunstig “hjernecelle” i netværket. I netværket strømmer information fra én ende til den anden gennem flere lag af neuroner. Hver neuron afgør, om den skal sende informationen videre, ved hjælp af en såkaldt aktiveringsfunktion – en “tænd/sluk”-mekanisme, der gør det muligt for netværket at lære komplekse mønstre i data.
Store AI-modeller som ChatGPT og billedgenkendelsesprogrammer består af milliarder af forbundne neuroner, der arbejder sammen for at løse komplekse opgaver. Selvom netværkene er store, bygger de på samme grundlæggende idé.
For at træne et neuralt netværk skal det “fodres” med data, som kan være alt fra tekst og billeder til lyd, video eller katalytiske materialer, afhængigt af opgaven. Under træningen vurderer netværket sin egen præstation ved hjælp af en “fejl-funktion”, der måler, hvor tæt netværkets svar er på det korrekte svar. Netværket justerer derefter sine forbindelser for at forbedre sig, og denne proces gentages igen og igen, indtil det opnår den ønskede nøjagtighed.
Katalytiske materialer
Katalytiske materialer
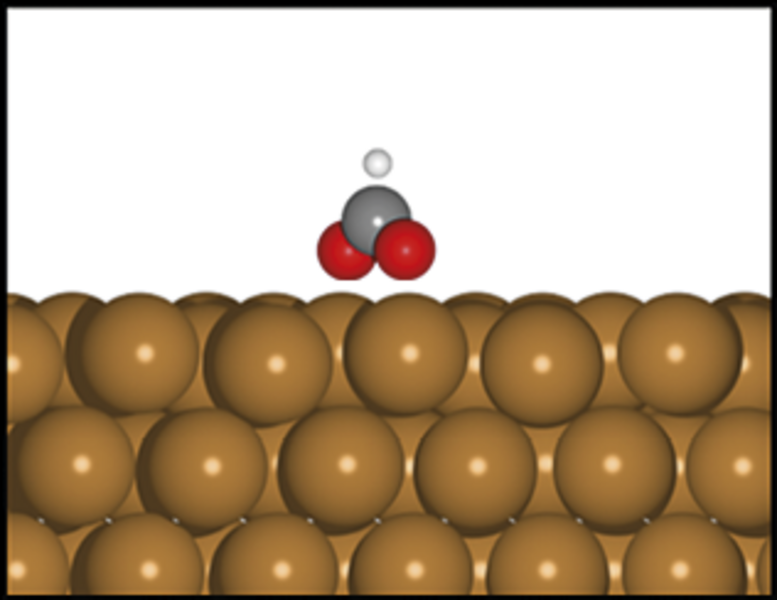
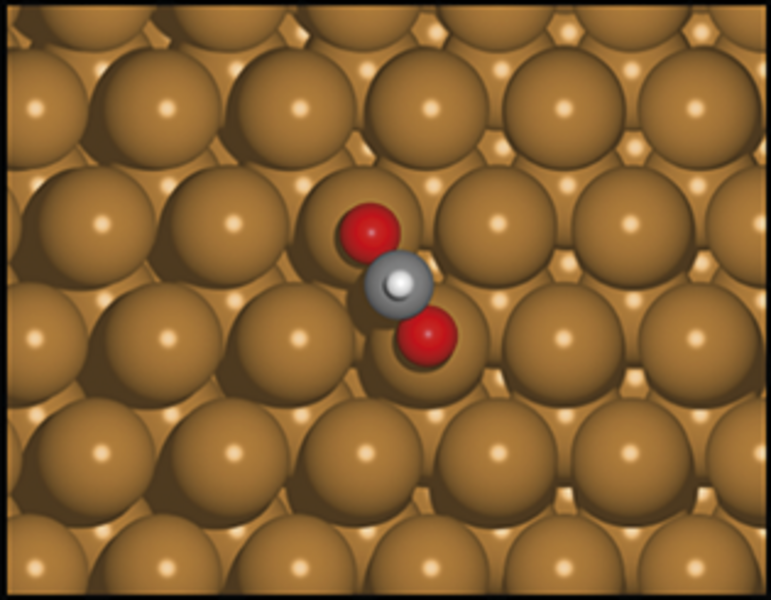
Et katalytisk materiale er et materiale, der kan øge hastigheden i en kemisk reaktion, uden selv at blive opbrugt eller omdannet i processen. Et godt eksempel er kobber, som kan bruges til at få carbondioxid, CO2, til at reagere med hydrogen, H2, og danne ønskede produkter, for eksempel metanol, CH3OH. Kobber kan altså hjælpe os med at reducere mængden af CO2 i atmosfæren og opnå et carbonkredsløb uden fossile brændstoffer ved at omdanne CO2 til et andet molekyle – metanol – som der er stor efterspørgsel på i den kemiske industri, og som også kan bruges som et syntetisk brændstof.
Reaktionen mellem carbondioxid og hydrogen (reaktanterne) sker på overfladen af det katalytiske materiale. Hvis reaktionen forsøges uden katalysator (ved blot at blande reaktanterne), er der en høj energibarriere. I praksis betyder det, at reaktionen tager så lang tid, at det ikke er økonomisk rentabelt. På overfladen af det katalytiske materiale kan energibarrieren sænkes ved at bryde den overordnede reaktion ned i små trin, hvor hvert trin har en lavere energibarriere.
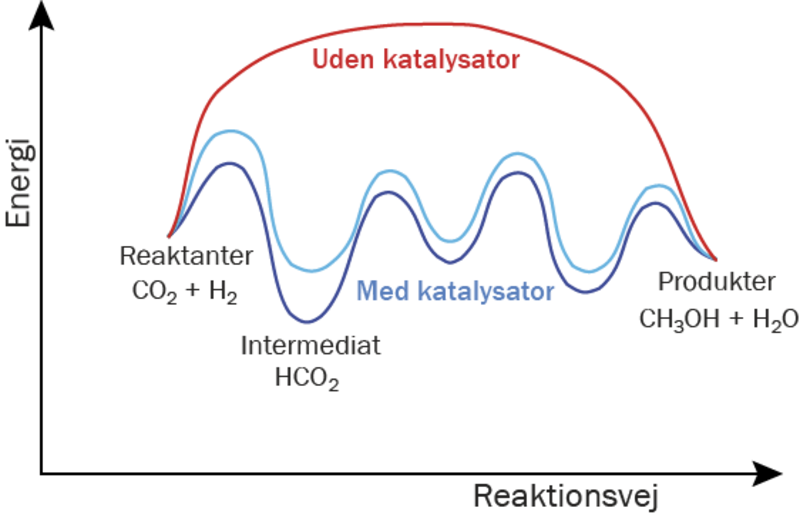
Et sådant trin er for eksempel dannelsen af molekylet format, HCO2, ved reaktionen mellem carbondioxid og et hydrogenatom. En vigtig parameter, der er afgørende for energibarrieren og dermed, hvor godt katalysatoren virker, er bindingsenergien – dvs. hvor stabil bindingen mellem intermediære molekyler som format og overfladen er. I forskning, hvor AI-modeller anvendes til at forudsige, om nye materialer er gode katalysatorer, fokuseres der derfor ofte på at forudsige bindingsenergier mellem molekyler og overflader. Bindingsenergien skal være netop tilpas, hverken for svag eller for stærk, for at overfladen er en god katalysator for dannelsen af det ønskede produkt.


Mie Andersen er lektor i teoretisk materialefysik. Hendes forskning har som mål at forstå og skræddersy katalytiske egenskaber af materialer – fra den atomare skala og op – ved hjælp af computer-modellering og AI-metoder.
mie@phys.au.dk
Nikolaj Rønne er postdoc i teoretisk materialefysik og forsker i udvikling af nye AI-metoder til materialeopdagelse på atomar skala.
nronne@phys.au.dk
Begge ved Institut for Fysik og Astronomi, Aarhus Universitet.