Biologisk kodebrydning - Kunstig intelligens fik has på proteinfoldningsproblemet
Proteiners funktion hænger nøje sammen med deres tredimensionelle struktur, som igen er dikteret af proteinets aminosyresekvens. At forudsige proteiners struktur ud fra deres aminosyresekvens har dog vist sig at være en næsten umulig nød at knække. Men årets nobelpristagere i kemi fik brudt koden.
Af Anna Ida Trolle og Kresten Lindorff-Larsen
Den 9. oktober i år er der en konference om proteiner på Københavns Universitet. Cirka kl. 12 går der et gisp gennem salen: Det annonceres, at Nobelprisen i kemi 2024 går til David Baker for databaseret proteindesign samt til Demis Hassabis og John Jumper for proteinstrukturforudsigelse. “De har knækket koden for proteiners fantastiske strukturer”, står der i pressemeddelelsen.
Men hvorfor skal vi være interesserede i, hvilke krøller og knuder der slås på de millioner af nanometer-store molekyler i dit, mit, din kats og din stueplantes indre?
I hverdagen møder du nok mest proteiner på forsiden af farverige indpakninger i supermarkedet, der, berettiget eller ej, praler af at være “high in protein”. Proteinet har indtaget scenen i supermarkedet, i fitnesscenteret og på sociale medier. Du er måske endda en smule træt af at høre om dem. Årets nobelpris er en perfekt anledning til at fortælle om, at proteiners rolle for liv ikke blot er en historie om æg, bønner og skyr.
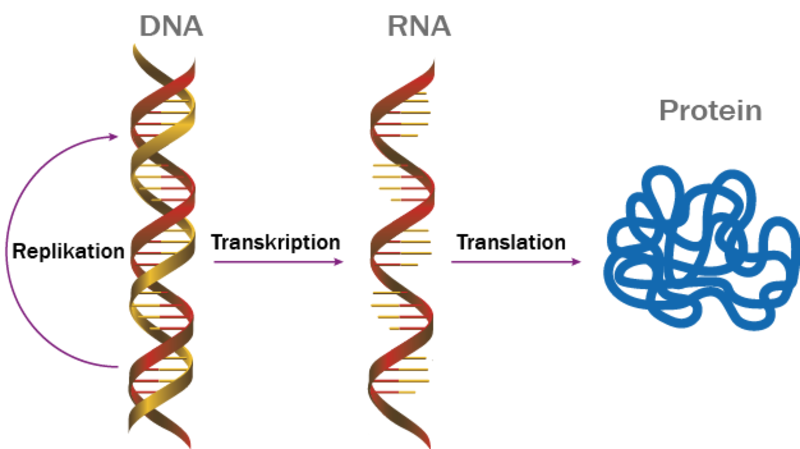
Når du trækker vejret, når bladene på træerne forgyldes og forgår, når du drømmer om natten, er det proteiner, der spiller en central rolle. Lange kæder af DNA indeholder opskriften på, hvilke proteiner der skal laves, og hvordan de skal se ud. Proteinerne er altså en slags molekylære maskiner, der indgår i et væld af biologiske processer.
Struktur og funktion hænger sammen
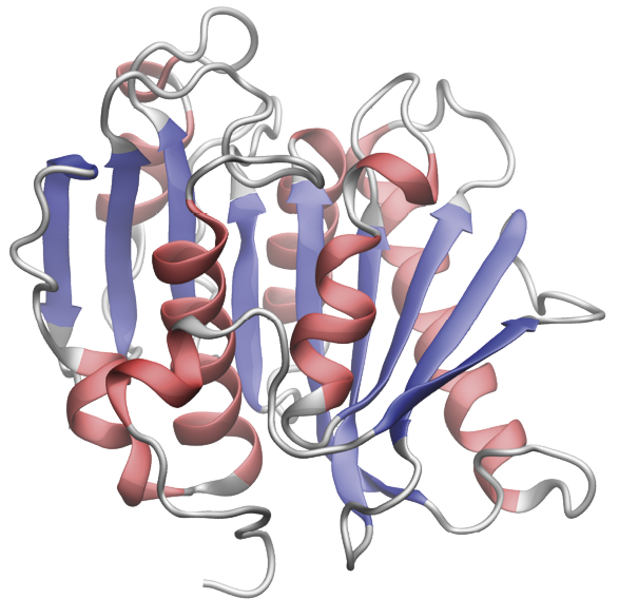
Proteiner kan have mange forskellige former og størrelser, og lige præcis deres struktur er interessant, fordi den er tæt forbundet med deres rolle i organismen. Når du bøjer din arm eller rynker på næsen, så er det lange filamenter af proteinerne myosin og aktin, der er i færd med at binde sig sammen, hvorefter myosin giver et ryk, der gør, at musklen sammentrækkes. Når du trækker vejret, transporteres oxygen rundt i din blodbane med proteinet hæmoglobin, som ligner en lille tornekrans, der kan fange oxygenmolekyler.
Trods milliarder af års evolution, der har udmøntet sig i en palet af vidt forskellige dyr, ligner hæmoglobinet i mennesker stadig til forveksling hæmoglobinet i alle andre dyr på jorden. I pattedyrs muskelceller findes også proteinet myoglobin, som ligeledes binder oxygen.
Myoglobin fungerer som et oxygenlager, og derfor har nogle dyr, såsom hvaler, en høj koncentration af myoglobin i musklerne, da de tilbringer en del tid under vandet. Både myoglobin og hæmoglobin binder også jern, hvilket er grunden til, at blod og rødt kød har en karakteristisk jernsmag.
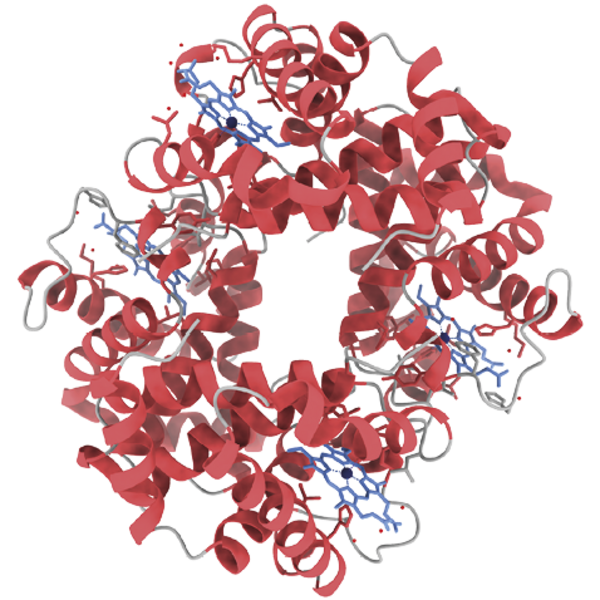
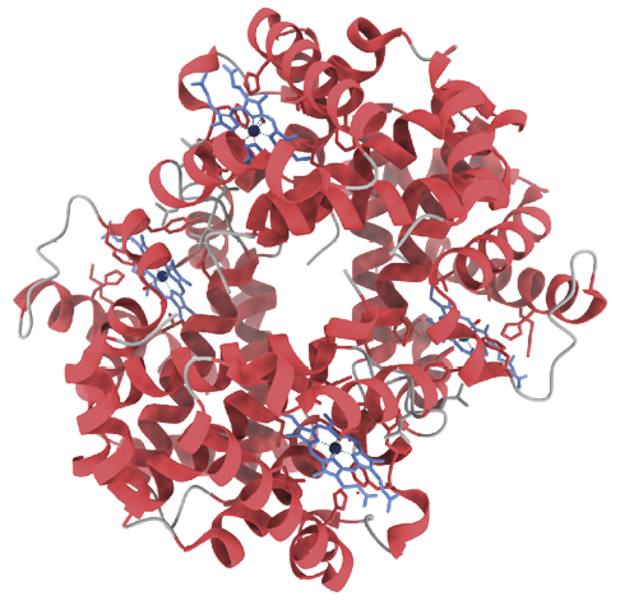
Hæmoglobins struktur er velkonserveret i pattedyr. Her ses hæmoglobin fra mennesker (venstre) og kvæg (højre). De blå molekyler er hæm, som koordinerer jernioner.
Det ihærdige makkerpar, myoglobinet og hæmoglobinet, bringer os ydermere tilbage til nobelprisen. I 1962 delte John Kendrew og Max Perutz nobelprisen i kemi for at have bestemt strukturen på henholdsvis myoglobin og hæmoglobin med røntgenkrystallografi, en metode der stadig er udbredt. Det er altså ingen gennemsnitlig bedrift at kortlægge hvert atom i et protein, ej heller i dag.
Små fejl kan give store problemer
Samtidigt med at detaljeret proteinstrukturbestemmelse var blevet muligt, begyndte forskere også at kaste sig over et andet problem: Hvad bestemmer, hvordan et protein ser ud?
Ligesom DNA er opbygget af en kæde af sammenbundne molekyler kaldet nukleotider, så er proteiner sammensat af en kæde af aminosyrer. Vi mennesker har 20 forskellige aminosyrer i vores repertoire, og med forskellige sammensætninger af disse i lange kæder dannes alt fra enzymer til antistoffer. Aminosyrer har forskellige egenskaber: Nogle har en elektrisk ladning, nogle er klistrede, nogle vil gerne interagere med vand, andre vil ikke.
Det viser sig, at når man placerer et proteins aminosyresekvens i et tilnærmelsesvist cellulært miljø, så vil det altid folde sig sammen til den samme, komplekse krølle. Et proteins struktur er altså fuldstændigt indkodet i dets aminosyresekvens. For denne opdagelse fik Christian B. Anfinsen i 1972, ti år efter Kendrew og Perutz, nobelprisen i kemi. Denne erkendelse er nu kendt som Anfinsens hypotese, og den er stadig central for moderne proteinkemi.
Sammenhængen mellem proteiners aminosyresekvens, struktur og funktion er et fascinerende molekylært puslespil. Men som det oftest er med biologi, er der en alvorlig skyggeside. Tilsyneladende små fejl i en DNA-streng kan føre til proteiner, der er formet forkert – eller slet ikke formet overhovedet. Cirka 1 ud af 25 mennesker i Nordeuropa bærer rundt på en særlig mutation. De mangler tre nukleotider i deres DNA. Mennesker har tre milliarder DNA-basepar, så en forskel på tre lyder måske ikke livstruende, men det er det.
Hvis begge forældre bærer denne mutation, er der cirka 25 % sandsynlighed for, at deres barn vil mangle en aminosyre på position 508 i begge af deres kopier af proteinet Cystic Fibrosis Transmembrane conductance Regulator (CFTR). CFTR understøtter transporten af vand og kloridioner i lungeceller. Manglen på aminosyre 508 medfører sygdommencystisk fibrose, og betyder, at der over tid opbygges slim i lungerne, hvilket fører til vejrtrækningsproblemer og gentagne lungeinfektioner. Den forventede levealder med cystisk fibrose er 40-50 år i Danmark. Vores forståelse af proteinstruktur er altså bogstaveligt talt livsvigtig.
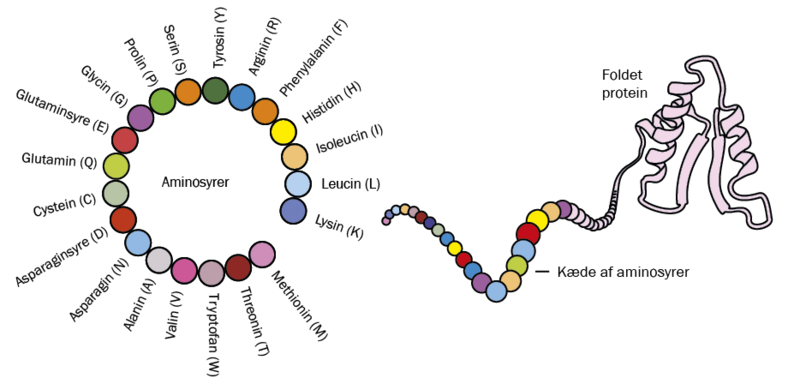
Et tilsyneladende uløseligt problem
Vi skal tilbage til Anfinsens hypotese. Hvis alle proteiners struktur er 100 % forudbestemt af deres aminosyresekvenser, så må det være muligt at forudsige et proteins 3D-struktur. Solen går ned over det 20. århundrede, computere bliver stærkere og erfaringerne større, men “proteinfoldningsproblemet”, som det blev kaldt, forblev uløst. Datamangel kan man ikke klage over: Siden 1971 er over 200.000 eksperimentelt bestemte proteinstrukturer blevet deponeret i databasen Protein Data Bank (PDB) sammen med deres aminosyresekvens.
Det er heller ikke af mangel på vilje eller anstrengelse; proteinernes grammatik er tåget og uklar, selv for avancerede AI-modeller, der kan finde mønstre i de mindste sammenhænge. Hvert andet år, i over 20 år, konkurrerer forskningsgrupper fra hele verden ved eventet CASP (Critical Assessment of Structure Prediction) i at skabe modeller, der kan løse proteinfoldningsproblemet. I 1994 kan den bedste model forudsige en proteinstruktur med cirka 48 % nøjagtighed, når man sammenligner aminosyrernes position i forhold til en eksperimentel struktur.
Eksperimentelle metoder som røntgenkrystallografi kan bryste sig med mere end 90 % nøjagtighed. I 2002 nåede man op på omkring 58 %, og ti år senere opnår den vindende model 59 % nøjagtighed. Der er lang vej til en nogenlunde pålidelig model, hvis hvert procentpoints forbedring kræver ti års benhård konkurrence mellem nogle af de bedste forskere i verden.
AlphaFold slår alle konkurrenter af banen
Endnu værre er det, hvis man kigger på modellernes præstation i at forudsige svære proteinstrukturer. CASP-konkurrencen har forskellige kategorier af sværhedsgrad. I den lette ende kan alle være med, alle modeller gennem årene kan forudsige en proteinstruktur med 80-90 % nøjagtighed. Men i den svære ende ligger forudsigelserne helt nede på mellem 20 og 40%.
Man kunne begynde at tro, at proteinfoldningsproblemet er uløseligt. Men i 2018 sker der et kvantespring. Googles AI-laboratorie, DeepMind, anført af Demis Hassabis og John Jumper, melder sin ankomst i konkurrencen med en model så overlegen, at den slår ned som et lyn fra en klar himmel. To år efter, til den 14. CASP-konkurrence i 2020, gør de det igen, og denne gang er deres model så god, at proteinfoldningsproblemet erklæres for løst.
Modellen kaldes for AlphaFold2, og det er for denne præstation, at Hassabis og Jumper modtager Nobelprisen i kemi i 2024. AlphaFold2 kan forudsige gigantiske proteinstrukturer helt ned til atomar præcision og er altså i mange tilfælde konkurrencedygtig med klassiske laboratoriemetoder, såsom røntgenkrystallografi. Forskellen er, at det kan tage årevis at kortlægge et protein i laboratoriet.
Med AlphaFold2 tager det kun få minutter, og det kan foretages af alle med en internetforbindelse. Det betyder, at hvis forskere kan nøjes med en god model, kan de springe det komplicerede laboratoriearbejde over. Og hvis de går i laboratoriet kan de bruge AlphaFold-modellen som hjælp i deres arbejde.
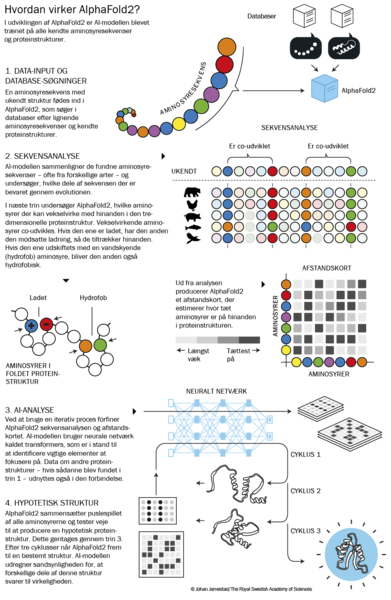
Fra proteinsfoldningsproblem til proteindesign

AlphaFold blev den endelige sammensvejsning mellem proteiners aminosyresekvens og struktur. Men hvis man kan forudsige proteinstruktur fra sekvens, er det så også muligt at gå den anden vej – altså udtænke et protein og så generere aminosyresekvensen, der skal til for at opnå en forudbestemt struktur?
Her kommer David Baker ind i billedet. Baker er, lige så vel som Jumper og Hassabis, en pioner indenfor proteinfoldning.
Hans bidrag til løsningen af proteinfoldningsproblemet er imponerende nok, men hans pragtpræstationer finder man inden for feltet proteindesign. Allerede i 2003 lykkedes han med at designe, og folde, et fuldstændigt nyt protein. I 2008 præsenterer han to nye enzymer for verden, og siden er tusindvis af nye proteiner materialiseret fra Bakers laboratorie. Bakers proteiner kan mere end at folde sig sammen til en forudbestemt tredimensionel struktur: De bruges i alt fx nanomaterialer, vacciner, og lægemidler.
Baker, Hassabis og Jumper fortjener Nobelprisen. David Baker er en af proteindesignets Grand Old Men, og Hassabis og Jumper er centrale figurer i AlphaFolds udvikling. Dog kan Nobelprisen og dens sagnomspundne uddelingsceremoni bidrage til en opfattelse af, at videnskabelige opdagelser er produktet af få, ensomme genier. Men vi må ikke glemme, at fremskridt, i særdeleshed i naturvidenskab, er resultater af et verdensomspændende, vedvarende samarbejde.
Artiklen, der først beskrev AlphaFold, blev publiceret i Nature i 2021 og har 34 forfattere. Machine learning-modeller, såsom AlphaFold, kan ikke blive bedre end det data, de er trænet på. Det har taget tusindvis af forskere millioner af timer at oprense og karakterisere de proteiner, der danner grundlag for modellens hidtil usete nøjagtighed. Det er altså ikke spildt arbejde, og ikke blot fordi mængden af data kan bruges til at træne AI.
Hjælp fra fysikkens love
Proteinfoldningsproblemet har i virkeligheden to facetter. Hverken AlphaFold eller David Bakers proteindesign besvarer centrale spørgsmål om, hvordan proteiner folder, som de gør. Mange sygdomme, herunder cystisk fibrose, opstår fra fejl i foldningsprocessen. Derfor er lyset stadig tændt i de mange laboratorier rundt omkring i verden, der forsker i biokemi.
De til stadighed åbne spørgsmål vedrører også især en gruppe proteiner, der slet ikke folder sig sammen i et ordnet garnnøgle, men i stedet veksler mellem et utal af strukturer. Disse “uordnede proteiner” udgør cirka en tredjedel af proteinerne i menneskekroppen og er især relevante for sygdomme som Alzheimers og Parkinsons. De er besværlige at studere i laboratoriet, og det betyder, at der er mangel på data, og at AlphaFold2 derfor ikke kan forudse deres strukturer. Vi må derfor gå anderledes til værks.
På Københavns Universitet arbejder vi med at kombinere eksperimenter med computersimuleringer. I stedet for at bruge kunstig intelligens, som betinger en stor mængde træningsdata, kan man i stedet antage, at aminosyrerne i en proteinkæde følger fysikkens love og derefter lade en computer udregne, hvordan proteinet vil foldes. Med denne metode har vi for nylig sammen med vores kolleger studeret alle uordnede proteiner hos mennesker og fundet nye sammenhænge mellem struktur og funktion.
Flere nobelpriser på højkant
De seneste 75 år har budt på enorme fremskridt indenfor både strukturel biologi og computervidenskab. Årets nobelpris i kemi afspejler, at kunstig intelligens ikke kun er til for, at ChatGPT kan lave dine lektier. Det er værd at nævne, at nobelprisen i fysik i år gik til John Hopfield og Geoffrey Hinton, der deler prisen for deres bidrag til feltet machine learning. Dette faktum, kombineret med at CASP-konkurrencen skulle afholdes i over to årtier inden et gennembrud, demonstrerer, at det altså ikke er lutter lagkage at udvikle machine-learning-modeller.
Næste gang du er ude at handle og ser en bøtte skyr eller en proteinbar, så lad det minde dig om, at der inde i dig foregår et dramatisk, men ordnet virvar af kemiske reaktioner mellem hundredtusindvis af molekyler. Vi forstår dem bedre nu, end vi gjorde før Baker, Hassabis og Jumper, men der er stadig nobelpriser på højkant. Og mon ikke flere af dem vil gå til forskning, der opstår i krydsfeltet mellem menneskelig og kunstig intelligens. ♦
Fold selv proteiner med AlphaFold
Fold selv proteiner med AlphaFold
Her kan du finde en beskrivelse af, hvordan du selv kan folde proteiner med ColabFold, som er en online-version af AlphaFold. Med ColabFold kan enhver bruge AlphaFold uden at installere hele AlphaFold-programmet på egen computer.
Alt, du skal bruge, er: En pc, en Google-konto og en internetforbindelse.
ColabFold er en online-version af AlphaFold. Med ColabFold kan enhver bruge AlphaFold uden at installere hele AlphaFold-programmet på egen computer.
Trin 1)
Klik på dette link, der tager dig til ColabFold:
https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb
Trin 2)
Klik på ‘Fil’ i øverste venstre hjørne og derefter tryk ‘Gem en kopi i Drev’. Dette laver en kopi af notesbogen på din egen Google-konto, så du kan gemme dine resultater. Du kan trygt lukke den fane, der popper op med ‘Produktbemærkninger’.
Trin 3)
Øverst ser du et felt der hedder ‘query_sequence:’. Her kan du indsætte din egen proteinsekvens, eller blot prøve med den sekvens der allerede står i feltet. Du kan fx. prøve med nogle af de følgende:
(NB: Samles til en linje uden linjeskift)
Hæmoglobin subunit alfa fra Homo sapiens (menneske):
MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPT
TKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPN
ALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPA
VHASLDKFLASVSTVLTSKYR
Hæmoglobin subunit alfa fra Physeter macrocephalus (kaskelothval):
VLSPADKTNVKAAWAKVGNHAADFGAEALERMFMSFPST
KTYFSHFDLGHNSTQVKGHGKKVADALTKAVGHLDTLPD
ALSDLSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPGDFT
PSVHASLDKFLASVSTVLTSKYR
Alpha synuclein fra Homo sapiens (et protein involveret i Parkinsons sygdom):
MDVFMKGLSKAKEGVVAAAEKTKQGVAEAAGKTKEGVL
YVGSKTKEGVVHGVATVAEKTKEQVTNVGGAVVTGVTA
VAQKTVEGAGSIAAATGFVKKDQLGKNEEGAPQEGILED
MPVDPDNEAYEMPSEEGYQDYEPEA
Trin 4)
Når du har indsat den sekvens du gerne vil forudsige strukturen af, skal du trykke oppe øverst på fanen ‘Kørselstid’ og så vælge ‘Kør alle’.
Du kan følge med længere nede på siden hvor der efter lidt tid vil dukke nogle strukturer op. Efter et par minutter vil du kunne se en færdig struktur under fanen ‘Display 3D structure’. Du kan tage fat i strukturen med din mus og dreje den rundt.
Strukturen er farvet efter dens ‘pLDDT score’, som beskriver hvor sikker AlphaFold er på dens egen strukturforudsigelse. Hvis du prøver både med hæmoglobin og med alpha synuclein, vil du bemærke, at hæmoglobin er helt mørkeblå: AlphaFold er meget sikker på, at denne struktur er den rigtige.
Alpha-synuclein er derimod farvet rød, gul, og grøn. Det er fordi, det er et ‘uordnet protein’, som AlphaFold ikke kan forudsige. Dem kan du læse lidt mere om i artiklen.


Anna Ida Trolle studerer biokemi på Københavns Universitet, hvor hun også forsker i proteiners struktur og dynamik.
anna.ida.trolle@bio.ku.dk
Kresten Lindorff-Larsen er professor ved Linderstrøm-Lang Centret for Proteinvidenskab, Biologisk Institut, Københavns Universitet. Han underviser og forsker i proteinstruktur, dynamik og design.
lindorff@bio.ku.dk